
Progress
The Centre for Rare Disease Diagnosis, Research and Training has developed clinical, genomic, and experimental resources to advance rare disease diagnosis and research in India. These efforts have led to the establishment of a large clinical cohort, a biorepository and patient registry, improved diagnostic yield through multi-omics approaches, and functional validation of disease-causing variants. The following sections highlight this progress, organized around the centre’s core objectives.
Establishing a local clinical cohort, biorepository, and a rare disease registry
Clinical Cohort :
To support systematic rare disease research, the centre has established a large and well-characterized clinical cohort.
- To date, 1,250 families have been recruited for genetic testing, comprising 399 consanguineous and 851 non-consanguineous families.
- Comprehensive genomic analyses have been undertaken across the cohort, enabling stratification of completed and ongoing cases, diagnosed and undiagnosed individuals, and candidate gene findings.
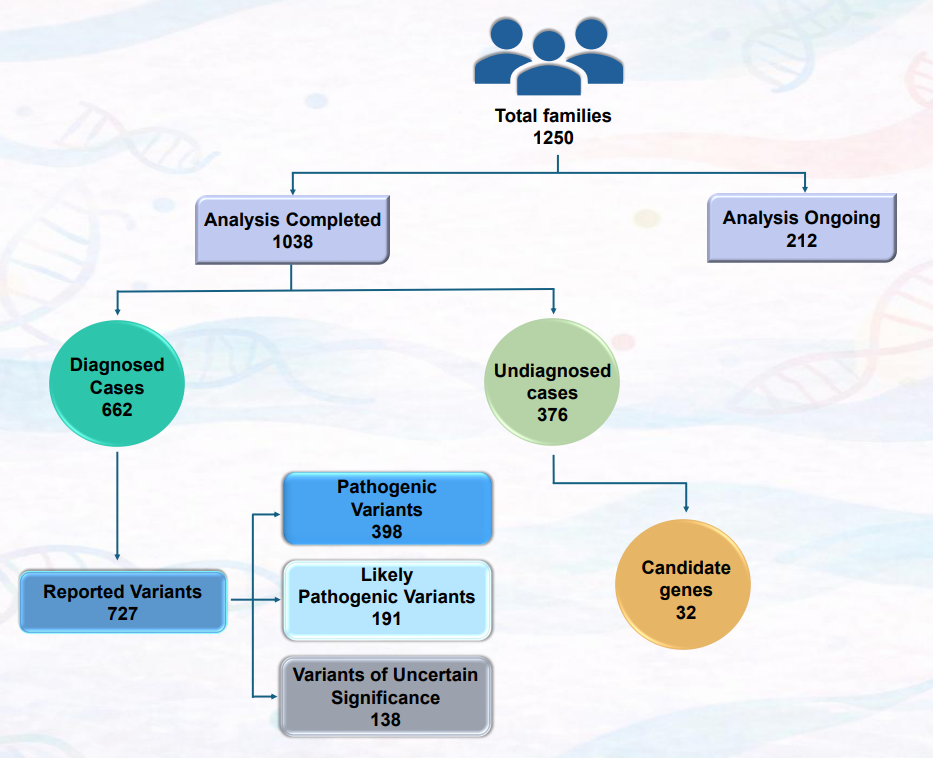
The schematic summarizes analysis status (completed vs. ongoing), diagnostic outcomes (diagnosed vs. undiagnosed), distribution of reported variants by ACMG classification, and candidate gene identification among 1,250 recruited families.
Biorepository :
Parallel to cohort development, a centralized biorepository has been established to support downstream molecular and functional studies.
- Over a five-year period, peripheral blood samples from 3,498 individuals belonging to 1,250 families (affected and unaffected members) have been collected.
- Genomic DNA has been isolated, standardized, and systematically banked for long-term use.
- In addition, 103 tissue samples from 88 affected individuals (from 87 families) have been collected and taken up for downstream functional studies.
- All biospecimens are barcoded, anonymized, and securely maintained within the centre’s biorepository.
Rare Diseases Registry :
To enable long-term data integration and collaborative research, the centre is developing a comprehensive rare disease patient registry.
- The registry is a secure, centralized platform that curates de-identified clinical and genomic data.
- Data captured include demographic details, clinical features, genomic findings, and variant classification.
- The registry is designed to support data sharing and multi-institutional collaboration, strengthening rare disease research in India and globally.

Building on this foundational infrastructure, the centre has applied advanced genomic and transcriptomic approaches to improve diagnostic resolution in rare genetic disorders.
Deploying Whole Exome Sequencing (WES), Whole Genome Sequencing (WGS), and RNA Sequencing (RNA-Seq) to discover potential disease-causing variants
A definitive diagnosis is the first step toward comprehensive care for individuals with rare genetic disorders. To maximize diagnostic yield, the centre has implemented a tiered, multi-omics diagnostic strategy combining genomic and transcriptomic approaches. A diagnostic yield of 63% have been achieved as of now.
Whole exome sequencing :
- WES was routinely employed as a first-tier test for a significant proportion of recruited individuals.
- Duo, trio, or quad exome sequencing was pursued when initial singleton analyses were inconclusive.
- To date, exome sequencing has been performed for 877 individuals to establish a definitive molecular diagnosis.
Whole genome sequencing :
WGS was employed for selected families in whom a definitive diagnosis could not be achieved through exome sequencing.
- WGS is employed for rare genetic disorders as it detects a wider range of pathogenic variants, including non-coding and structural variants.
- We have completed whole genome sequencing for 50 individuals belonging to fifty different families.
The following is an example of an individual in whom WGS was useful in prioritizing the disease-causing variants.
Impaired SERPINF1 expression due to c.[-37C>A];[829_831del] causes osteogenesis imperfecta VI
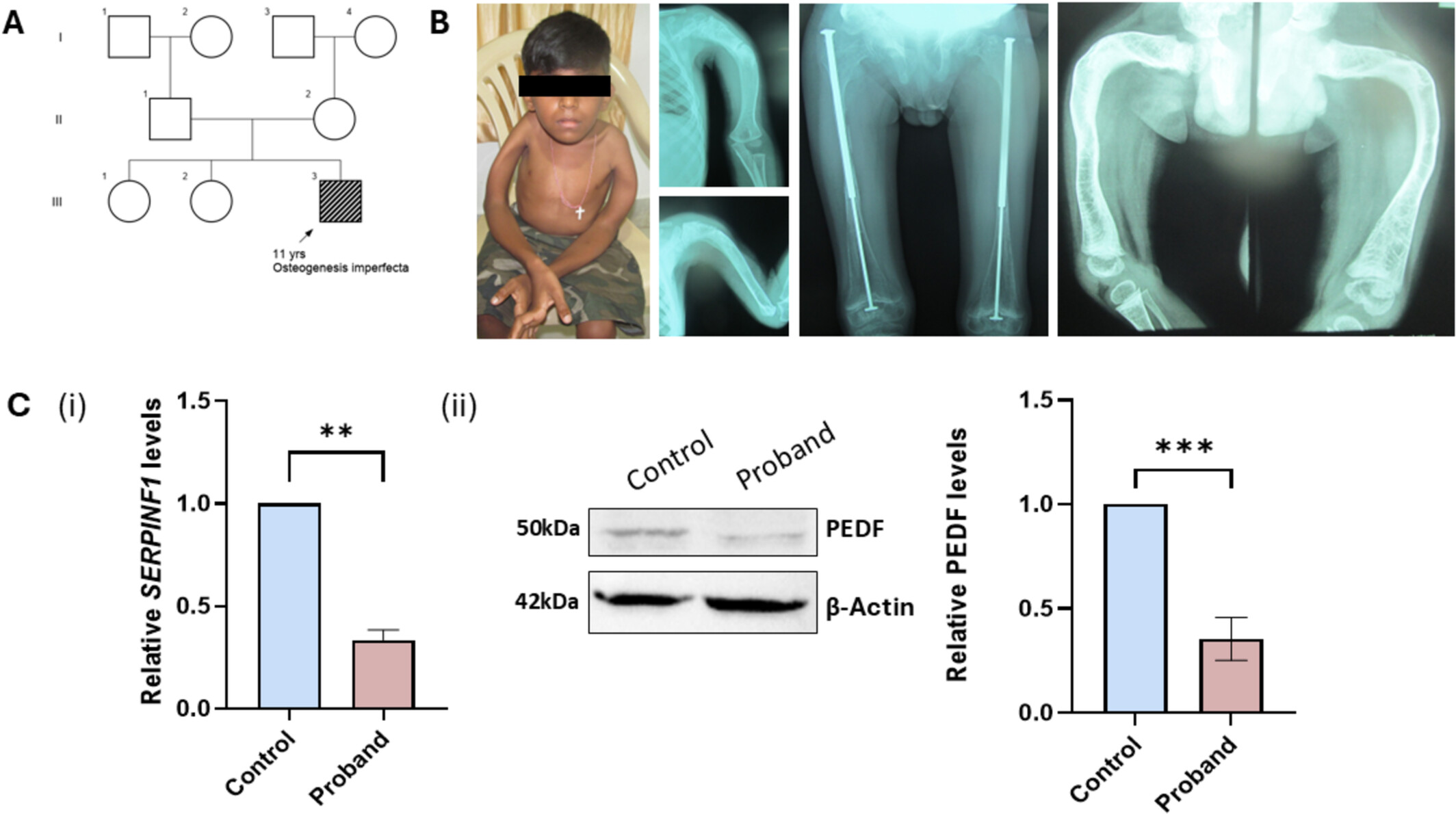
An eleven-year-old boy, born to a non-consanguineously married couple (A), presented with multiple fractures. Radiographic examination showed bilateral bowing of long bones, severe osteopenia, and vertebral deformities (B). WES analysis revealed compound heterozygous variants, c.[-37C>A];[829_831del] in SERPINF1, causing osteogenesis imperfecta type VI. Furthermore, WGS identified no other potential causative variants in the intergenic or deep intronic regions of SERPINF1. Functional analysis performed in patient-derived fibroblasts illustrated reduced expression of SERPINF1 transcript and protein levels (C.i, C.ii).
Read More: https://doi.org/10.1002/ajmga.70020
RNA Sequencing :
Transcriptomics has been used as a complementary approach alongside WES/WGS for prioritizing potential disease-causing variants and to re-classify the variants of uncertain significance, thereby improving the diagnostic yield in individuals with a possible rare genetic disorder.
In this project, transcriptomic analysis was specifically applied to unresolved cases, including individuals with variants of uncertain significance and those who remained undiagnosed.
- RNA-seq was performed for 17 families, selected based on gene–variant characteristics, expression profiles, sample availability, and funding.
- Among these,
- 7 families had no clinically relevant variants identified by DNA sequencing,
- 4 families harbored variants in genes of uncertain significance (ANKRD44, LCTL, ANKMY1, GABPA), and
- 6 families carried variants of uncertain significance in known disease-associated genes (PRIM1, GALNS, F5, UNC13D, NFATC2, BRCA1).
The following is an example where utility of RNA-seq helped in resolving a variant of uncertain significance.
A deep intronic variant, c.121-210C>T results in an in-frame 159bp pseudo-exon inclusion in intron 1 of GALNS
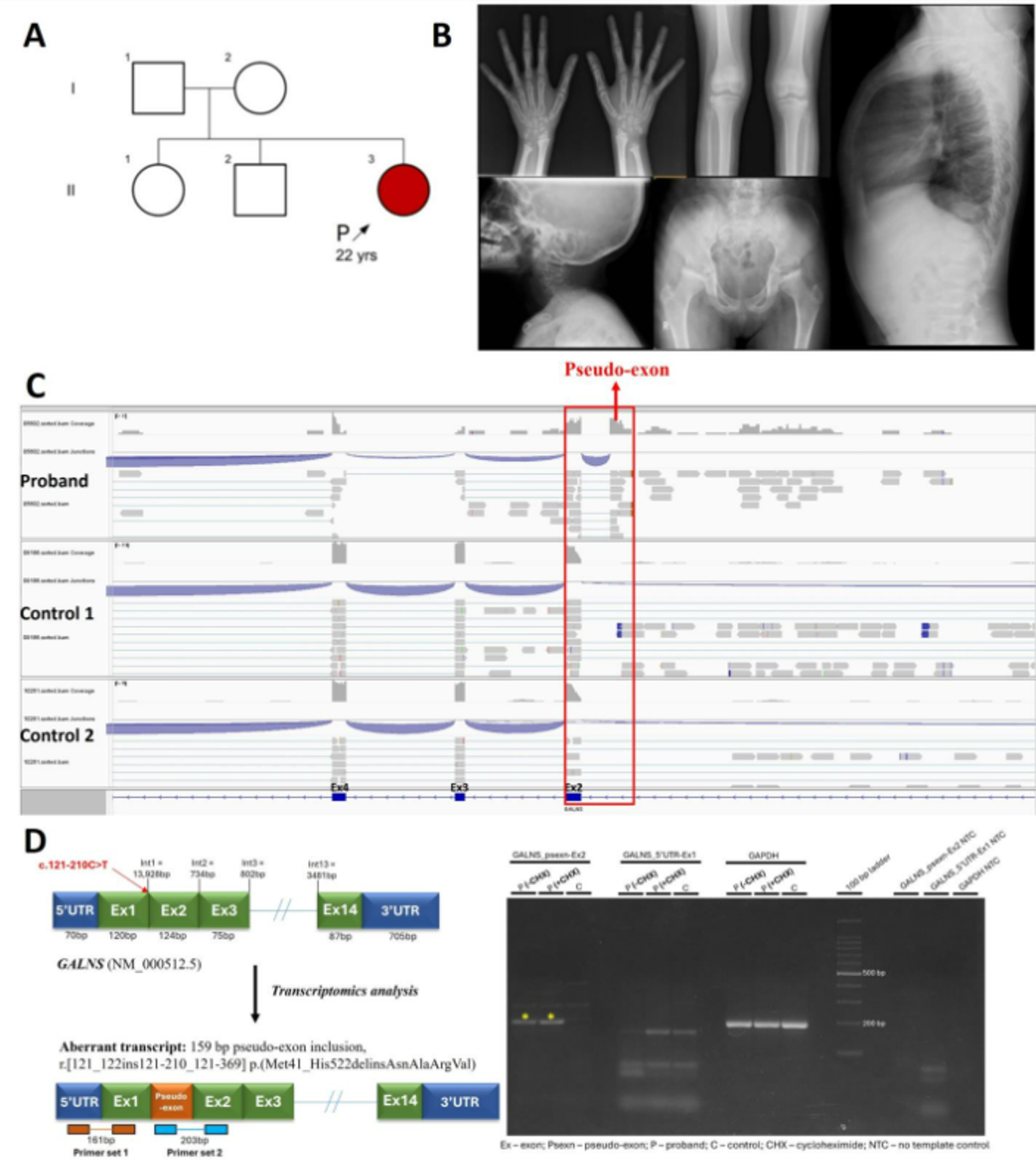
We ascertained a 23-year-old female with short stature, hip and ankle pain on prolonged standing and diffuse corneal clouding (A). X-ray survey revealed bilateral shortening of distal end of ulna, collapse of bilateral capital femoral epiphysis and biconvexity of lumbar and thoracic vertebral bodies (B). Singleton exome sequencing revealed a homozygous deep intronic variant, c.121-210C>T in intron 1 GALNS (NM_000512.5), which causes mucopolysaccharidosis IVA (MIM #253000, autosomal recessive). SpliceAI predicted that this variant leads to the formation of a new donor site with a delta score of 0.63 and an acceptor site with a delta score of 0.31. However, the variant is present in the population database gnomAD (v4.1.0) in 36 homozygotes. To determine the pathogenicity of the variant, transcriptomics was carried out using patient-derived fibroblasts. On analysis, we observed there was a significant reduction in the GALNS transcript levels (log2 fold change = -3.02; p-value = 0.0001; FDR = 0.103) compared to five controls. In addition to the reduction in transcript levels, we observed a new splicing event in the intron 1 region, indicating a pseudo-exon inclusion (C). These findings were validated by RT-PCR using cDNA derived from cycloheximide treated and untreated patient fibroblasts (D).
Functional characterization of causal genes and variants in cells and model organisms
The widespread availability and application of next-generation sequencing have enabled comprehensive genetic testing in individuals with rare disorders. While this has significantly improved diagnostic capabilities, it has also led to an increasing number of variants of uncertain significance (VUS) and the identification of novel gene–disease associations that require functional validation to establish clinical relevance.
To address this challenge, the centre employs an integrated functional genomics framework combining in-vitro cellular assays, proteomics, metabolomics, lipidomics, and animal models to validate candidate variants and elucidate disease mechanisms. The choice of experimental model is guided by the gene of interest and the biological pathways involved.
The following schematic illustrates genes harboring variants of uncertain significance that were prioritized for functional validation using the aforementioned approaches.

The center has been able to perform the validation of variants of uncertain significance from different genes using in-vitro functional studies on patient fibroblasts, proteomics and relevant animal models for variants.
The following study demonstrates the use of proteomics and zebrafish models to delineate the functional consequences of MRPS2 variants, leading to oxidative phosphorylation (OXPHOS) deficiency and early developmental abnormalities.
Further delineation of defects in MRPS2 causing human OXPHOS deficiency and early developmental abnormalities in zebrafish
MRPS2 (MIM*611971) encodes mitochondrial ribosomal protein S2, a core component of the small mitochondrial ribosomal subunit. Biallelic variants in MRPS2 cause combined oxidative phosphorylation deficiency 36 (COXPD36; MIM#617950), a primary mitochondrial energy failure disorder resulting from impaired mitochondrial ribosome function and defective respiratory chain biogenesis.
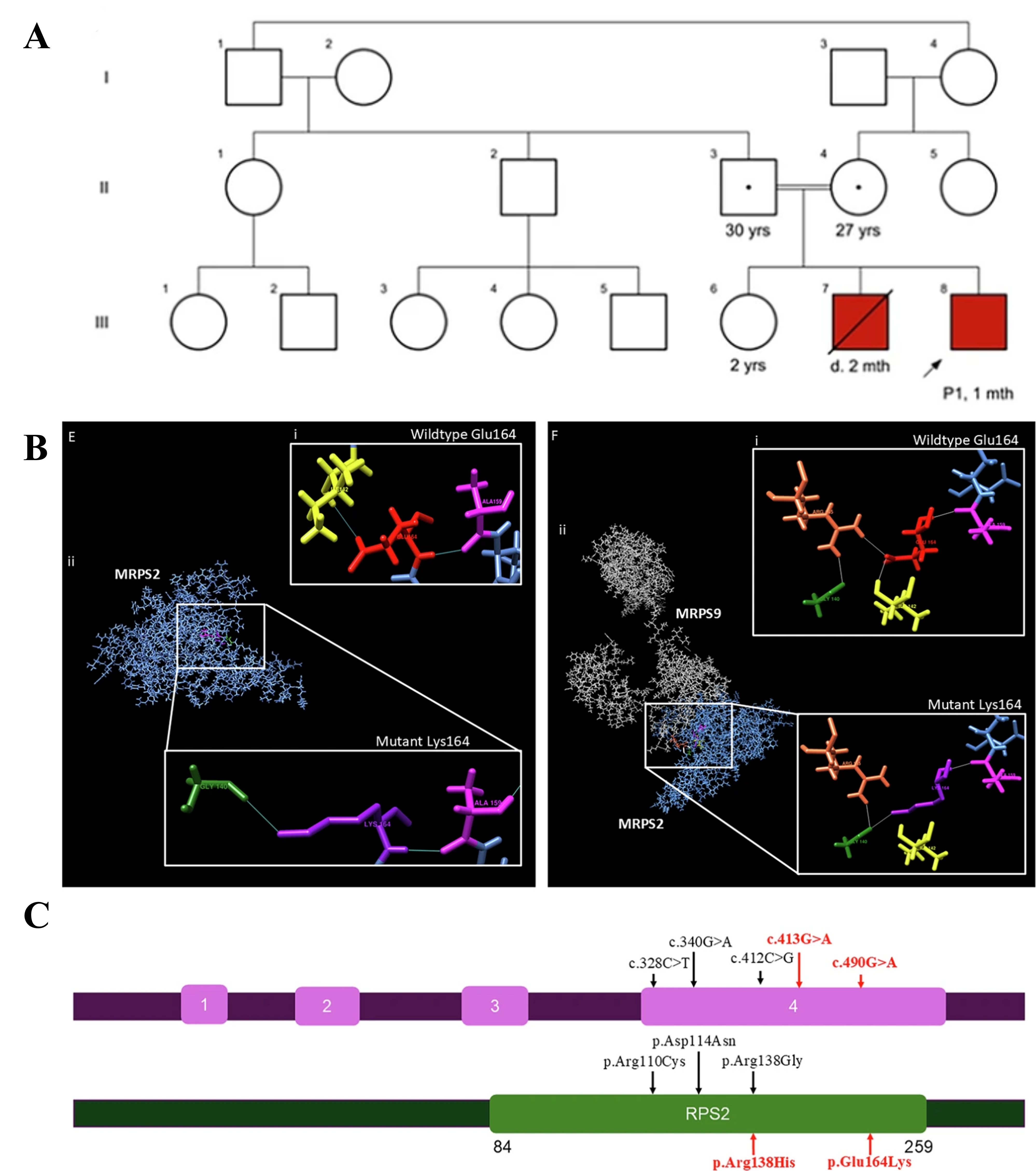
Figure 1: (A) Pedigree of family (B) In-silico protein modelling of the MRPS2 structure showing the c.490G>A p.(Glu164Lys) variant. (C) Schematic representation of MRPS2 (NM_016034.5) and protein showing our variants (highlighted in red) and previously reported variants. RPS2: Ribosomal protein S2.
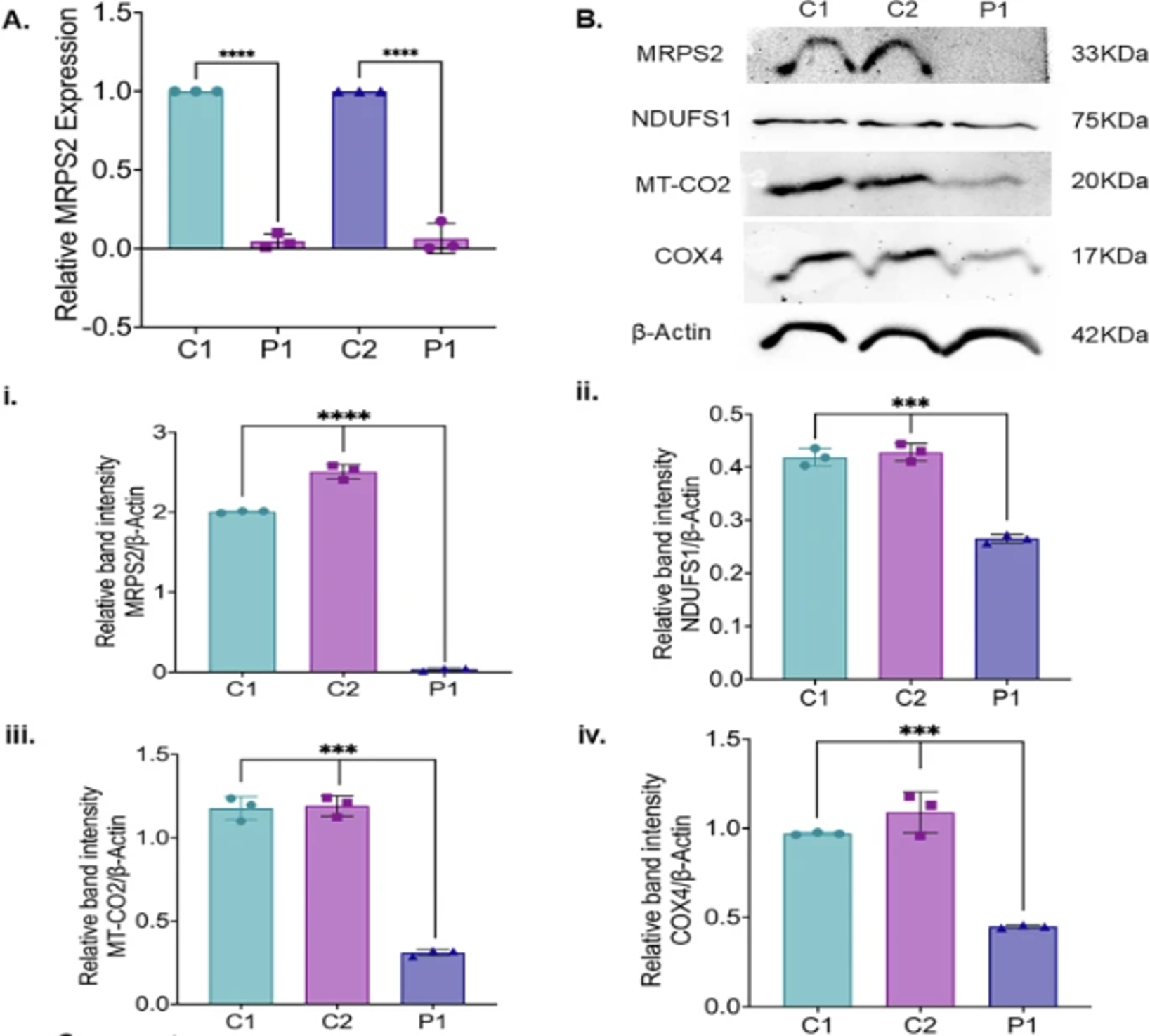
Figure 2: (2A) A qRT-PCR analysis showing relative MRPS2 mRNA expression in control 1 (C1), control 2 (C2) and patient (P1) cell lines. Expression of MRPS2 in P1 was significantly upregulated when compared to C1 and C2. β-Actin was used as the internal control; (2B) Representative blot images for protein expression of MRPS2, NDUFS1, MT-CO2 and COX4; (i-iv) Western blot analysis of protein expression of MRPS2, NDUFS1, MT-CO2 and COX4 in C1, C2 and P1 fibroblast cell lines. Densitometric analysis was performed upon normalization of MRPS2, NDUFS1, MT-CO2 and COX4 protein band intensity to respective β-Actin band.
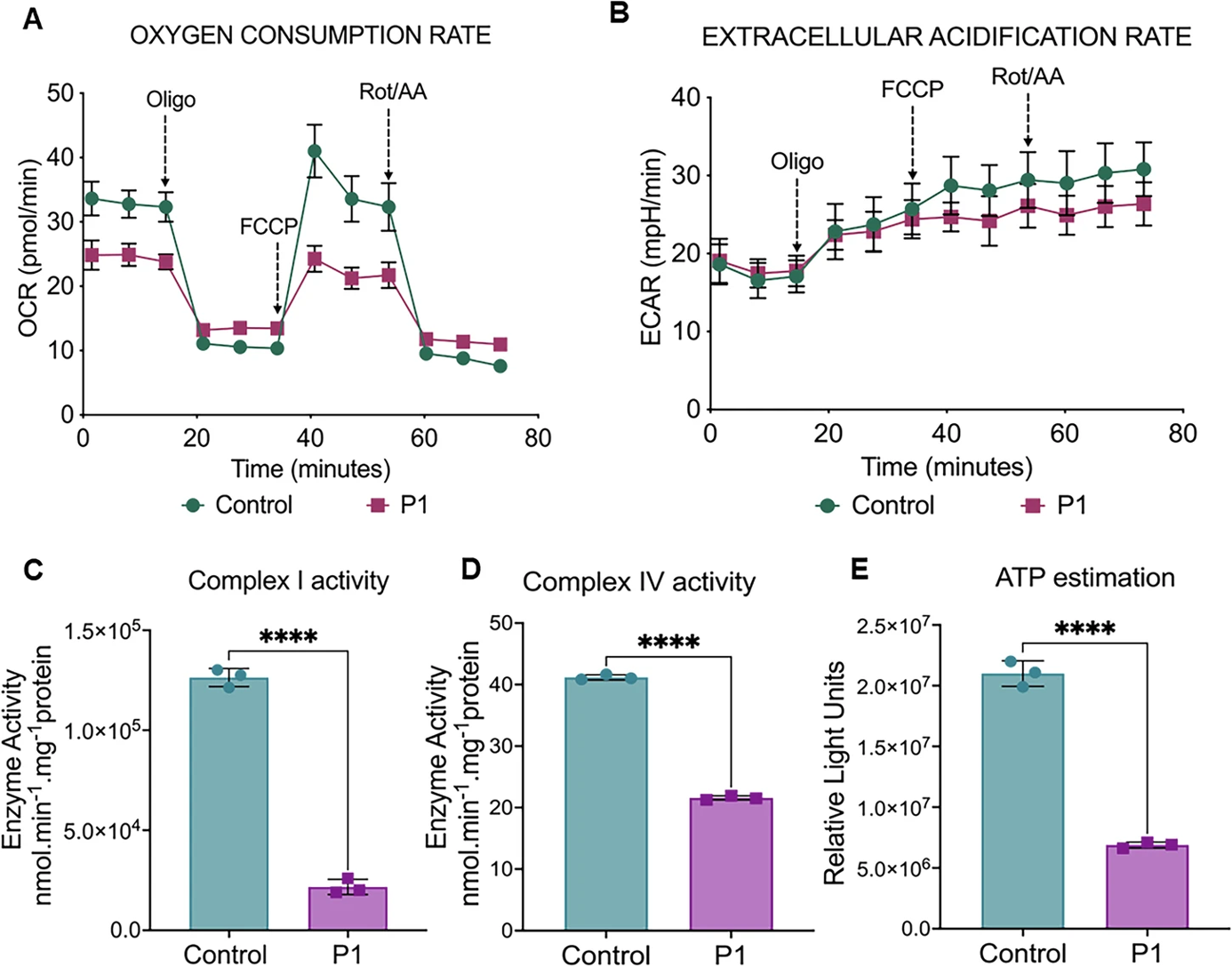
Figure 3: (A) Representative graph showing the OCR in control and P1 fibroblast cell lines. Injection of oligomycin, FCCP, and antimycin A and rotenone (A/R) are indicated (B) Representative graph showing the EACR in control and P1 cell lines. Injection of oligomycin, FCCP, and antimycin A and rotenone (A/R) are indicated (C) Mitochondrial complex I enzymatic activities in MRPS2 control and P1 cell lines (D) Mitochondrial complex IV enzymatic activities in control and P1 cell lines (E) Representative graph showing intracellular ATP levels. ATP determination kit was used to quantify intracellular ATP levels. *p < 0.05, **p < 0.001, ***p < 0.0001 and ****p < 0.00001.
The functional impact of the c.490G>A p.(Glu164Lys) variant in MRPS2 was studied in patient-derived fibroblasts and controls which showed evidence of mitochondrial dysfunction. The proband demonstrated reduced MRPS2 mRNA and protein levels, along with decreased OXPHOS complex I (NDUFS1) and complex IV (COX4/MT-CO2) subunits. Functional assays revealed impaired respiration with low oxygen consumption rate, reduced complex I and IV activity, and decreased ATP production. Imaging demonstrated abnormal mitochondrial structure and a reduced mitochondrial membrane potential, supporting a severe defect in mitochondrial energy metabolism.
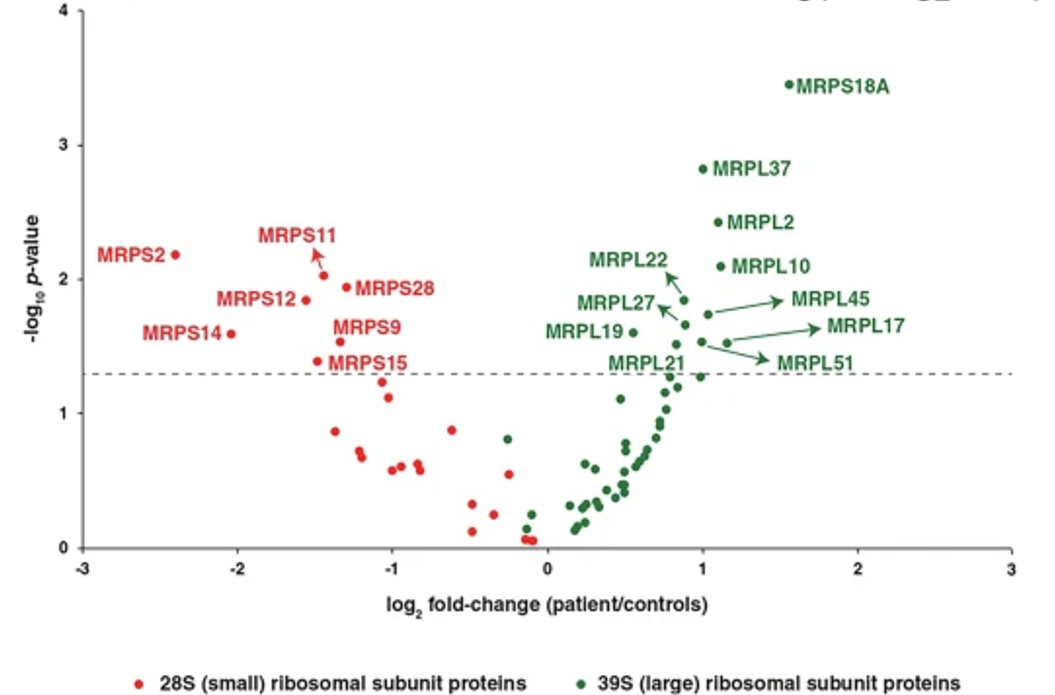
Figure 4: Altered abundance of mitochondrial ribosomal proteins. A volcano plot showing alterations in the levels of various mitochondrial ribosomal subunit proteins. Small and large mitochondrial ribosomal subunit proteins are represented by red and green dots, respectively, as indicated. The horizontal dashed line corresponds to p = 0.05.
On proteomics analysis, a significant decrease in MRPS2, NDUFS1, and COX4 expression was noted in proband when compared to controls.
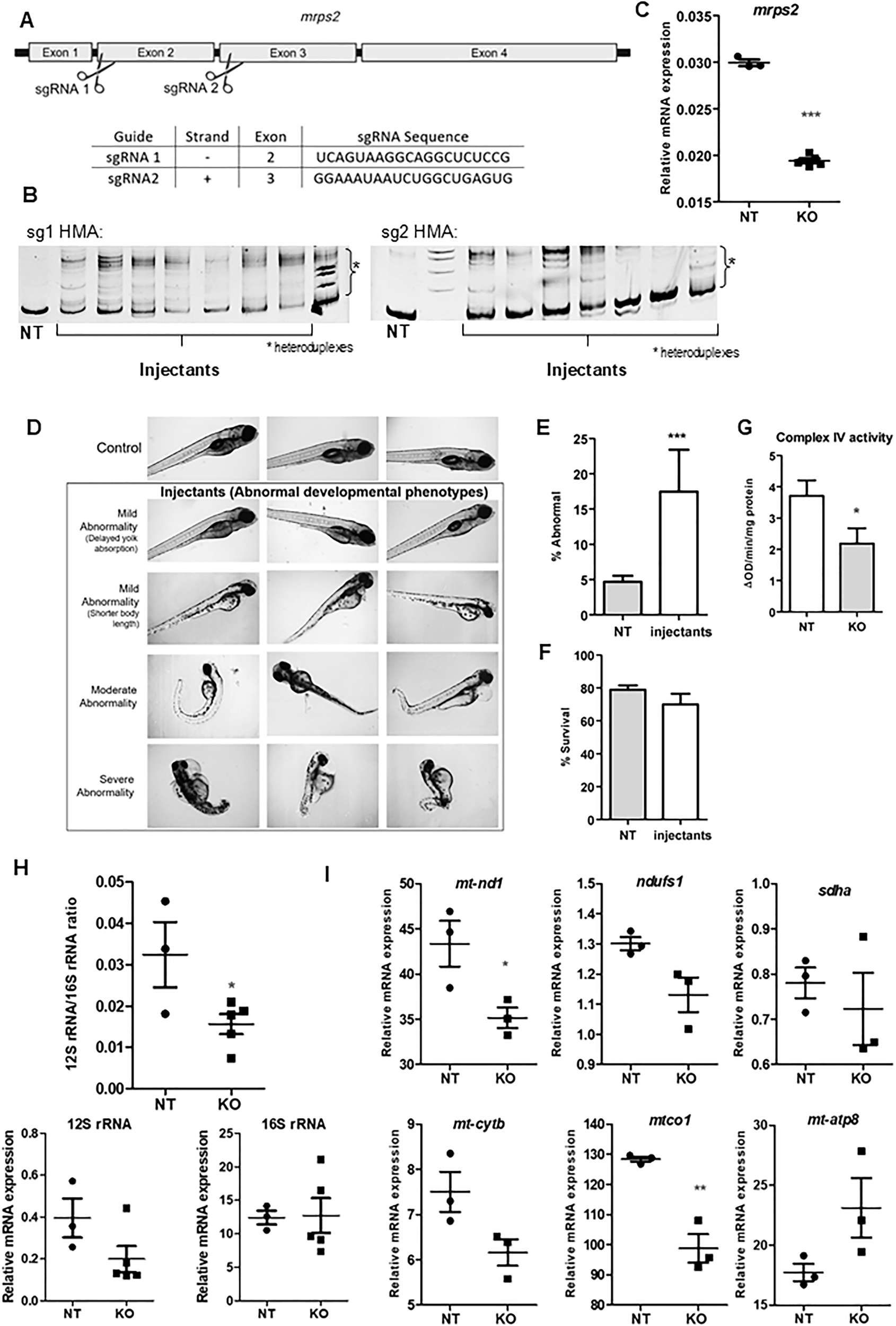
Figure 5: (A) Representative bright field images of wild-type and mrps2 crispants (at 3dpf) illustrating abnormal developmental phenotypes observed in the crispants, as labeled, (B-C) Quantification of the number of larvae showing abnormal development at 5dpf and survival at 7dpf (5 experiments, NT n = 560, KO n = 820), (D) Complex IV activity measurement in mitochondria isolated from control and mrps2 crispants, (E) 12S to 16S rRNA ratio in the crispants compared to controls. Results from 3 to 5 independent experiments are quantified, and error bars represent SEM, (F) Relative mRNA levels of OXPHOS subunits genes including nd1, ndufs1 (Complex I), sdha (Complex II), cytb (Complex III), mt-co1 and mt-co2 (Complex IV), in the mrps2 crispants compared to controls.
Additionally, mrps2 knockout zebrafish larvae exhibited an abnormal developmental phenotype despite preserved overall survival, together with reduced complex IV activity, decreased 12S rRNA levels, and lowered expression of mitochondrial respiratory chain complex I, IV, and V subunits, indicating defective small mitochondrial ribosomal subunit integrity and secondary impairment of oxidative phosphorylation.
These findings align with prior reports of impaired small subunit assembly in MRPS2-deficient cells and support a role for MRPS2 loss in altered mitochondrial ribosome composition, resulting in combined oxidative phosphorylation deficiency and a primary mitochondrial energy failure phenotype.
Read More: https://doi.org/10.1038/s41431-025-01858-1
CCN2 represents another example where functional validation was undertaken to establish its role in skeletal development.
Biallelic variants in CCN2 underlie an autosomal recessive kyphomelic dysplasia
Using CRISPR-Cas9 gene editing, we generated ccn2a knockout zebrafish (F0 crispants) and observed striking phenotypes: altered body curvature, bent or missing tails, and disrupted craniofacial cartilage formation. Alcian blue staining revealed underdeveloped cartilage structures such as Meckel’s cartilage and ceratohyal arches, while molecular assays showed reduced expression of key skeletal markers including col2a1a (chondrocytes), sp7 and runx2a (osteoblasts), and rhoAa (palatogenesis). These findings directly linked CCN2 loss of function to impaired chondrocyte and osteoblast differentiation.
By integrating clinical genetics with zebrafish functional studies, we demonstrated how biallelic CCN2 variants cause kyphomelic dysplasia. Zebrafish provided a rapid and powerful system to connect genetic discoveries in patients with mechanistic insights in vivo, underscoring the strength of collaborative research in bridging rare disease genomics with experimental biology.
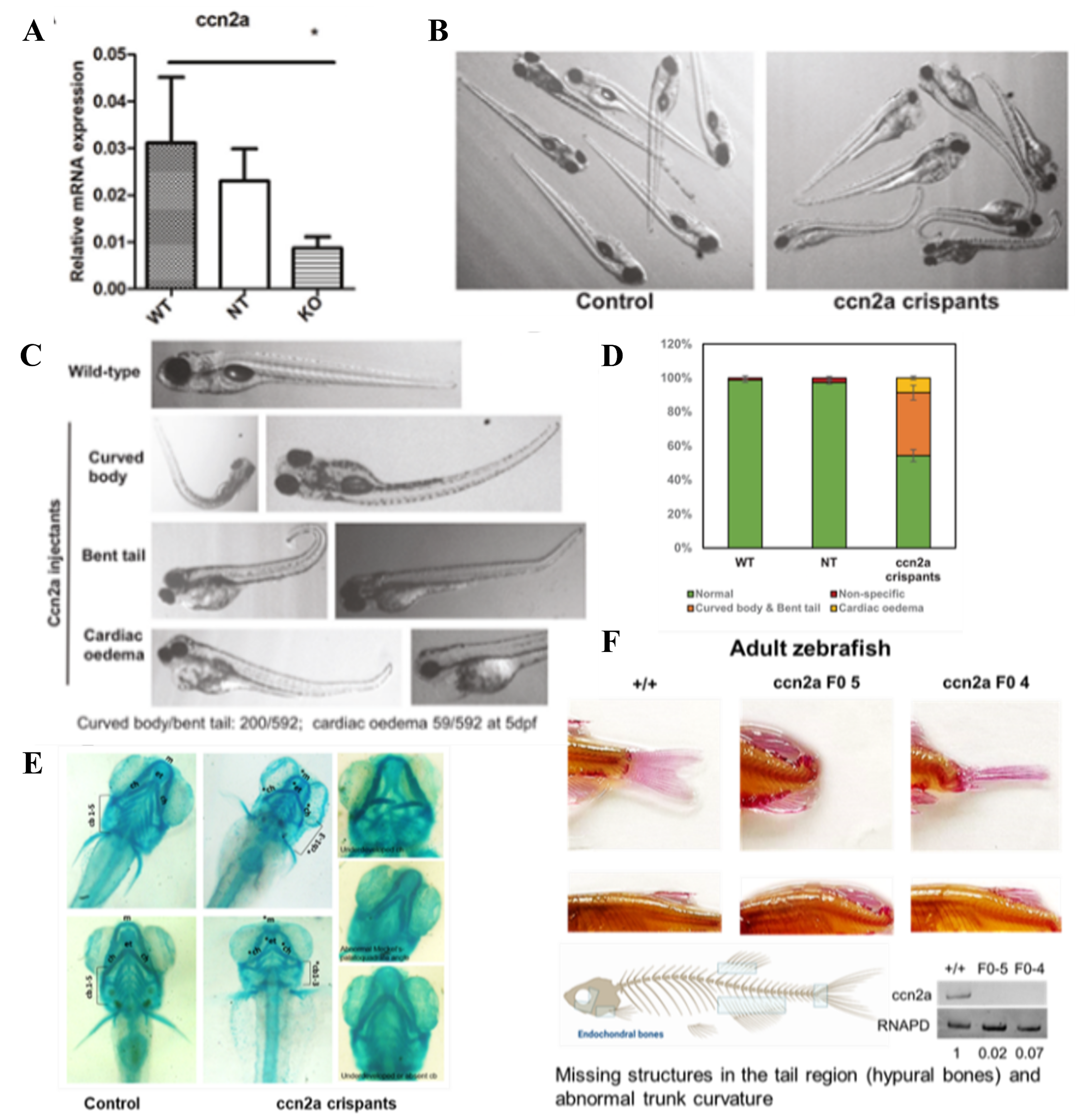
Phenotypes in the ccn2a FO knockout zebrafish. (A-D) Reduction in CCN2 protein levels using a CRISPER-Cas9 mediated gene editing strategy, results in abnormal development of zebrafish larvae with morphological abnormalities as shown. (E) Significant cartilage defects at 6.5dpf: Bent, missing or misshapen cartilage framework in the absence of CCN2, could be the basis for craniofacial defects. (F) Missing structures in the tail region (hypural bones) and abnormal trunk curvature.
Read More: https://doi.org/10.1038/s41431-024-01725-5
Data Sharing and Public Accessibility
The data generated from the work so far would aid the scientific community working with rare genetic disorders. The following data has been submitted to public repositories and is freely accessible:
- Genomics data:
Approximately 600 genomic variants, along with curated clinical information, have been submitted to the openly accessible ClinVar database.
(https://www.ncbi.nlm.nih.gov/clinvar/submitters/505991/)
- Zebrafish functional data:
Functional validation data generated using zebrafish models have been deposited in the ZFIN database
(ZDB-CRISPR-250206-1, ZDB-CRISPR-250206-2)
- Proteomics data:
Proteomic datasets generated during functional characterization studies have been deposited with the ProteomeXchange Consortium via the PRIDE repository
(PXD055397, PXD044775)
Through these submissions, the centre contributes high-quality, curated datasets to the scientific community, promoting transparency, reproducibility, and collaborative discovery in rare disease research.